EU MDR: European Union Medical Device Regulation of 2017
The Medical Device Regulation (EU) 2017/745, or the European Union Medical Device Regulation (EU MDR), is legislation that establishes quality and safety requirements for medical devices produced or supplied in the EU market. It combines legislation for medical devices and active implantable medical devices into one document.

The EU MDR replaces the Medical Device Directive (MDD) and Active Implantable Medical Device Directive (AIMD). Because the EU MDR is legislation in the form of a Regulation rather than a Directive, it is directly applicable at a European Union level without requiring additional local legislation.
The EU MDR changes the European Union’s legal framework for medical devices, greatly increasing the rigor and robustness of the regulations. The Regulation places restrictions and reporting requirements on substances used in the design and manufacturing of medical devices. It governs the production and distribution of medical devices in Europe and requires medical device companies that want to sell their products in the European marketplace to comply with the EU MDR. In addition, manufacturers must develop EU MDR-compliant regulatory systems, processes, and documents to continually monitor the safety and performance of their medical device products.
Let’s jump in and learn:
Who Must Comply with the EU MDR?
Any medical device company that wants to sell its products in the EU marketplace must comply with the EU MDR. This includes new devices and existing devices. Even if a device was certified under the MDD, it must comply with the rules set forth by EU MDR.
To prove compliance, every device must have a CE Mark or Conformitè Europëenne Mark, the European Union’s (EU) mandatory conformity marking for regulated goods sold within the European Economic Area (EEA). The CE Mark represents a manufacturer’s declaration that products comply with the EU’s New Approach Directives, including EU MDR.

The requirements for CE Marks are based on the class of the medical device. The level of risk that the medical device presents determines its classification, which directs the conformity assessment route for the device.
EU MDR Class I—Low Risk
EU MDR Class 1 is for the lowest risk medical devices. Included in EU MDR Class I are non-sterile devices or devices without a measurement function. Examples of EU MDR Class 1 medical devices are wheelchairs, glasses, and stethoscopes. Most medical devices in Class 1 can be self-assessed.
All other EU MDR medical device classes require a conformity assessment by a Notified Body and a CE Mark.
EU MDR Class I Special Function—Low/Medium Risk
There are a few EU MDR Class 1 exceptions that must have a notified body’s certification. EU MDR Class 1 special function medical devices include reusable surgical instruments, devices that are supplied sterile, and devices that have a measuring function.
EU MDR Class IIa—Medium Risk
EU MDR Class 2a includes invasive devices intended for short-term use. Examples of EU MDR Class 2a medical devices are tooth implants, tracheotomy tubes, syringes, X-ray devices, and surgical clamps.
EU MDR Class IIb—Medium/High Risk
Invasive devices intended for long-term use are in EU MDR Class 2b. Medical devices in Class 2b include implantable birth control, blood bags, and bone fixation plates.
EU MDR Class III—High Risk
High-risk, EU MDR Class 3 covers implantable medical devices intended for long-term use. EU MDR Class 3 devices include pacemakers, spinal disc implants, drug-coated stents, and heart valves.
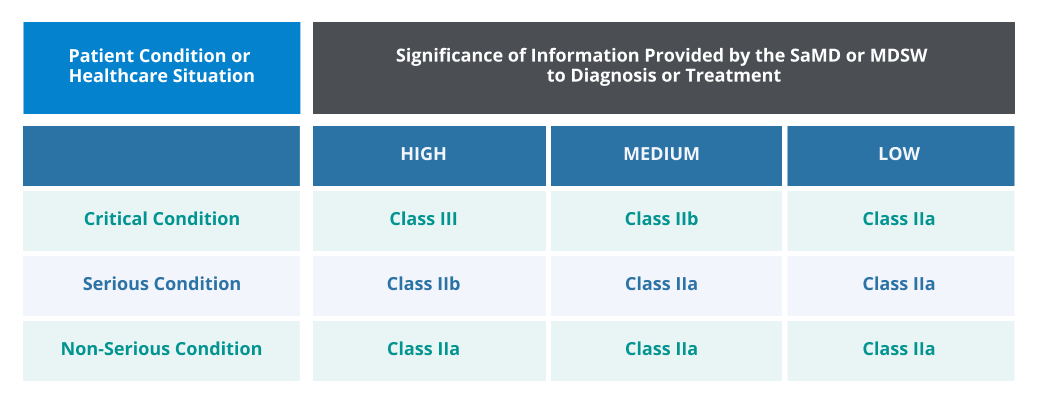
Software as a Medical Device (SaMD) or Medical Device Software (MDSW)
EU MDR compliance is required for digital products that meet the definition of software as a Medical Device or Medical Device Software. Rule 11 of EU MDR categorizes the types of software and the corresponding classifications.
SaMD or MDSW that must comply with EU MDR includes medical apps, Artificial Intelligence (AI) diagnosis and diagnostic-assist technology, medical imaging analysis software, decision support tools, and other software solutions with a medical purpose.
Legislation and Timeframes
The EU MDR replaces the existing Directives for medical devices (93/42/EEC and 90/385/EEC). It entered into force on May 26, 2017. EU MDR became fully applicable on May 26, 2021.
EU MDR Timeline
- May 2017: MDR published and in effect
- April 2020: Deadline for EU MDR compliance extended one year
- May 2021: CEs only issued to medical devices that adhere to EU MDR requirements
- May 2024: MDD certificates void, existing certified medical devices must be transitioned to meet MDR compliance requirements
New UDI Requirements
- May 2021: Class III medical devices and Implantable Devices
- May 2023: Class IIa and Class IIb products
- May 2025: Class I products
- Two years after the respective product class data: Reusable Products where the UDI must be placed on the product itself
EU MDR Challenges
The changes to medical device regulations that come with EU MDR present manufacturers with a number of challenges and increase their compliance burden. Among the EU MDR challenges are:
- Broader scope for medical device classification
Includes more products than MDD, such as medical software applications, contact lenses, and equipment used for liposuction - Enhanced requirements for clinical evidence
Includes all available clinical evidence (i.e., favorable or unfavorable), evidence collection according to EU MDR protocols, and real-world evidence (RWE) - Reclassification and up-classification of devices
New classifications based on the contact duration, risk, and invasiveness - Increased clinical testing requirements
Reassessment of clinical data for reclassified devices that are already on the market and clinical testing data required for devices newly classified to be under EU MDR - More demand for Notified Bodies
Notified Bodies must evaluate the medical devices for compliance before CEs can be issued, which, due to the reduction in authorized Notified Bodies, creates a bottleneck for approvals - Emphasis on post-market surveillance
Proactive monitoring of device performance for recertification, annual safety updates for higher-risk class devices, and rapid reporting of safety incidents - Mandatory Unique Device Identification (UDI)
UDIs are required to allow manufacturers and authorities to track specific devices through the supply chain - Person Responsible for Regulatory Compliance (PRRC) required
Manufacturers must identify a “qualified person” within their organization to be the PRRC who is responsible for all aspects of EU MDR compliance
Key Aspects of the EU Medical Device Regulations
Other key aspects of the EU medical device regulations that should be noted include:
- Serious consequences for non-compliance, including losing the CE mark, having to withdraw products from the EU market, and delaying launches of new devices
- Updates to the General Safety and Performance Requirements (GSPR) that require manufacturers to demonstrate that the device is “state of the art” as well as how the device meets GSPR, including general requirements, design and manufacture, and information supplied with the device
- Increased pre-market assessment for high-risk devices by an independent expert panel operated by a Notified Body
- Significantly enhanced requirements for clinical studies and the clinical data that is made available to the public and healthcare professionals
- More specific and increased requirements for the designation and ongoing monitoring of Notified Bodies across the EU to increase consistency and oversight efficacy
- Independent EU assessment of Notified Bodies candidates
- More detailed information about economic operators throughout the supply chain, including distributors and importers
- Comprehensive EU database of medical devices and device traceability based on UDI
Notified Body Considerations
EU MDR Notified Bodies are organizations designated by the EU Member State to assess medical devices and associated technical documents for conformity with the requirements of the EU MDR. When a Notified Body determines that a device submission is in conformity, it can issue a certificate of conformity. This allows the medical device manufacturer to affix a CE mark to the device and take it to the EU market. Manufacturers can engage any authorized Notified Body to assess their product.
Notified Bodies authorized to perform conformity assessments under MDD are required to be reassessed, which has resulted in a significant reduction in authorized organizations. In addition, Article 35 of the EU MDR requires that any EU Member States that wants to establish MDR-certified Notified Bodies must have an authority in place to oversee the activities of authorized Notified Bodies in their country.
Capabilities and Requirements for EU MDR Notified Bodies
- Offer their conformity assessment services to any manufacturer inside or outside the EU
- Operate in a non-discriminatory, transparent, neutral, independent, and impartial manner
- Have a team with sufficient knowledge and experience to carry out the conformity assessment in accordance with applicable product Class requirements
- Maintain confidentiality of all information obtained in the course of conformity assessments
- Provide assessment information to relevant notifying authorities, the market surveillance authorities, and other Notified Bodies
Conformity Assessment
A manufacturer can only place a product on the EU market when it meets all the applicable requirements. The conformity assessment procedure is carried out before the product can be sold. The European Commission’s main objective is to help ensure that unsafe or otherwise non-compliant products do not find their way to the EU market.
What conformity assessment is:
- The conformity of a product is assessed before it is placed on the market
- It needs to demonstrate that all legislative requirements are met
- It includes testing, inspection and certification
- The procedure for each product is specified in the applicable product legislation
European MDR FAQs
What is the European Medical Device Regulation (EU MDR)?
The Medical Device Regulation (EU) 2017/745 or the European Union Medical Device Regulation (EU MDR) is a regulation that establishes quality and safety requirements for medical devices produced or supplied in the EU market.
Does the EU MDR supersede other EU medical device rules?
Yes, the MDR supersedes the Medical Device Directive (MDD) and the Active Implantable Device Directive (AIMD).
Does the EU MDR only apply to companies based in the EU?
Yes, any medical device manufactured or supplied in the EU must comply with EU MDR.
What are the compliance requirements For EU MDR?
There are a number of requirements for complying with EU MDR, including:
- A CE Declaration of Conformity
- Adhere to strict labeling requirements
- Meet standards for storage and transport conditions
- Ensure traceability with UDIs and submission to EU database (EUDAMED)
- Provide detailed clinical evidence proving safety
- Perform ongoing, post-market surveillance
What is the EUDAMED?
EUDAMED stands for European Databank on Medical Devices. It provides centralized storage for information about medical devices. It is equivalent to the FDA’s MAUDE (Manufacturer and User Facility Device Experience).
The database includes UDI registration requirements and registration of other information, including medical device post-market follow-up, safety and clinical information, and manufacturer’s registration. The objectives of the EUDAMED are to increase transparency, ensure traceability of medical devices, and facilitate the flow of information between manufacturers and users of medical devices, Notified Bodies, EU member states, and the European Commission.
What changes for Notified Bodies under the EU MDR?
The notification procedures and the procedures for monitoring Notified Bodies have been enhanced, making them more strict. There are also changes that harmonize the rules across all EU states. In addition, Notified Bodies are required to employ more physicians or clinical experts and regularly rotate auditors.
What are unannounced audits?
EU MDR requires Notified Bodies to conduct unannounced audits at least once every five years. These audits include the inspection of products.
Which products are covered by the EU MDR?
All medical devices that previously fell within the scope of the Medical Device Directive (MDD, 93/42/EEC) and all products regulated under the previous Active Implantable Medical Device Directive (AIMDD, 90/385/EEC) are covered by EU MDR. Additional medical devices are included under EU MDR, such as products used to clean, disinfect, or sterilize medical devices, colored contact lenses, reusable surgical instruments, cosmetic devices, and devices for the prediction and prognosis of diseases and health conditions.
Will there be any changes to the classification of my medical device?
The previous system remains in place, with classification for most products remaining the same. A few changes include the addition of a new Class I product, reusable surgical Instruments, and the expansion of the definition of medical devices to cover new products, such as software, and some products without medical purposes, such as contact lenses.
A Higher Bar for Compliance Further Enables Patient Safety
The EU MDR raises the bar for medical devices manufactured and sold in the EU. The requirements for EU MDR compliance are more specific and strict than its predecessors MDD and AIMD, increasing the compliance burden on medical device manufacturers. However, the EU MDR harmonizes requirements across the EU and provides long-term patient safety benefits.
Egnyte has experts ready to answer your questions. For more than a decade, Egnyte has helped more than 16,000 customers with millions of customers worldwide.
Last Updated: 2nd December, 2021